This server is simple to use.
As an example you can just paste the sequence of the Bacitracin Peptide
Synthetase I (BacA) into the main window and
click ![]() .
.
TUTORIAL FOR THE PKS/NRPS ANALYSIS WEB SERVER |
This tutorial will highlight the main features of this webserver. The main goal is to provide you with a comprehensive overview. NRPS/PKS peptide sequence files have been provided to use in this tutorial: Click on the link below, which will open in a separate window, copy and paste the sequence in the main page window to follow the example. |
This server is simple to use.
As an example you can just paste the sequence of the Bacitracin Peptide
Synthetase I (BacA) into the main window and
click
|
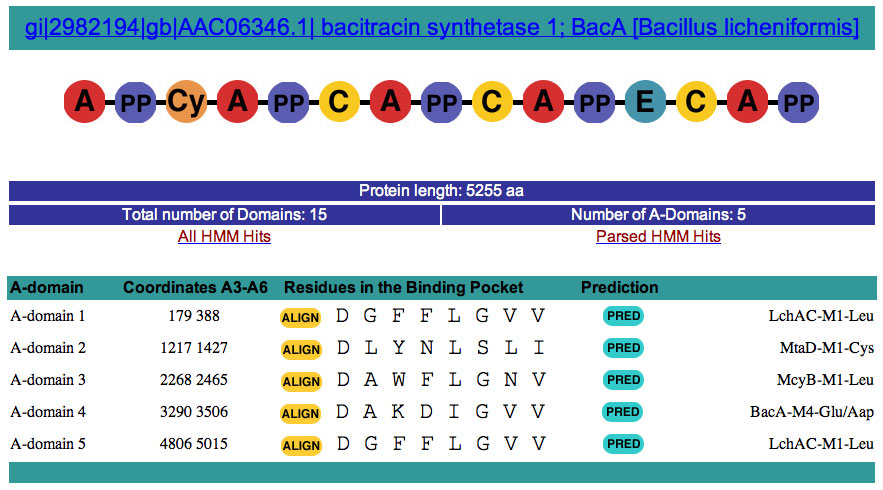
This is a snapshot of the output for BacA.
|
Clickable Objects: 1. By Clicking on the Name of the protein you just analyzed you obtain the sequence of the entire protein in fasta format.
2. By Clicking on a Domain you obtain the sequence and the coordinate in fasta format: For example for the first A-domain
>A_DOMAIN_1 14..538
KMTENEKELILHFNNTKTDYPKNKTLHELFEEQAMKTPDHTALVFGAQRMTYRELNEKAN
QTARLLREKGIGRGSIAAIIADRSFEMIIGIIGILKAGGAYLPIDPETPKDRIAFMLSDT
KAAVLLTQGKAADGIDCEADIVQLDREASDGFSKEPLSSVNDSGDTAYIIYTSGSTGTPK
GVITPHYSVIRVVQNTNYIDITEDNVILQLSNYSFDGSVFDIFGALLNGASLVMIEKEAL
LNINRLGSAINEEKVSVMFITTALFNMIADIHVDCLSNLRKILFGGERASIPHVRKVLNH
VGRDKLIHVYGPTESTVYATYYFINEIDDEAETIPIGSPLANTSVLIMDEAGKLVPIGVP
GELCIAGDGLSKGYLNREELTAEKFIPHPFIPGERLYKTGDLAKWLPDGNIEFIGRIDHQ
VKIRGFRIELGEIESRLEMHEDINETIVTVREDEESRPYICAYITANREISLDELKGFLG
EKLPEYMIPAYFVKLDKLPLTKNGKVDRKALPEPDRTAGAENEYE
3. By Clicking on the COMPUTER PREDICTION: D G F F L G V V as seen below. BLAST ALIGNMENT FOR A-domain 1 4. By clicking on the Query= AD1 5. HMM all Hits/HMM parsed Hits This server uses Hidden Markov Model (HMM) to predict the identity of each domain in this multi-modular enzymes. Hidden Markov Models are statistical
representations of groups of proteins which share sequence, and consequently,
functional similarity. HMMs were built for each known domains from NRPS and PKS. Each HMMs is tested against a larger set of protein to determined the lowest score possible that will still identify the specific domain the HMM was built against. This score is call the "trusted" score or cut-off. A specific cut-off has been determined for each HMMs. By clicking on the " HMM All Hits" link you can see all the HMM hits before these cut-off score are applied to the entire set of HMM Hits. This is what you get after for example for BacA.fasta LIST OF PARSE HMMs HITs for gi|2982194|gb|AAC06346.1| bacitracin synthetase 1; BacA [Bacillus licheniformis] A_DOMAIN 1/5 179 388 .. 1 228 [] 357.6 2.9e-107
What you see here for example. A_DOMAIN 3/5 2268 2465 .. 1 228 [] 347.4 3.3e-104 A_DOMAIN is the name of the HMM for A-domains. 3/5 means that it is the 3rd hit out of five for A-domains. 2268 2465 is the coordinates on BacA of the Hit. The A-domain HMM was built using only the A3-A6 sequence this is what you see here. .. Means that the hit was good at both the Nterminus and Cterminus. If a part of the HMM is missing you will see something like that [.which indicates that the N-terminus is missing or had a very low score, but the C-terminus is present with a high score. 347.4 is the actuall HMM score and is above the trusted cut-off in this case as it is in the parsed HMM hits list. 3.3e-104 represent the probability that this hit is to a A-domain. In this case really good.
In this list, by looking at the coordinates of each domains you can very quickly see if there were domains that the analysis didn't predicted or a with novel function. This is also done automatically if the gap between two domains is more than 450 aa. This is very arbitrary, but it serves well in the testing phase. In the case an unknown domain is found, this is the icon you will see in the top part of the output screen:
In addition, a message will appears at the bottom of the screen indicating the possibility of a unrecognized domain and it's coordinates on the protein.
|